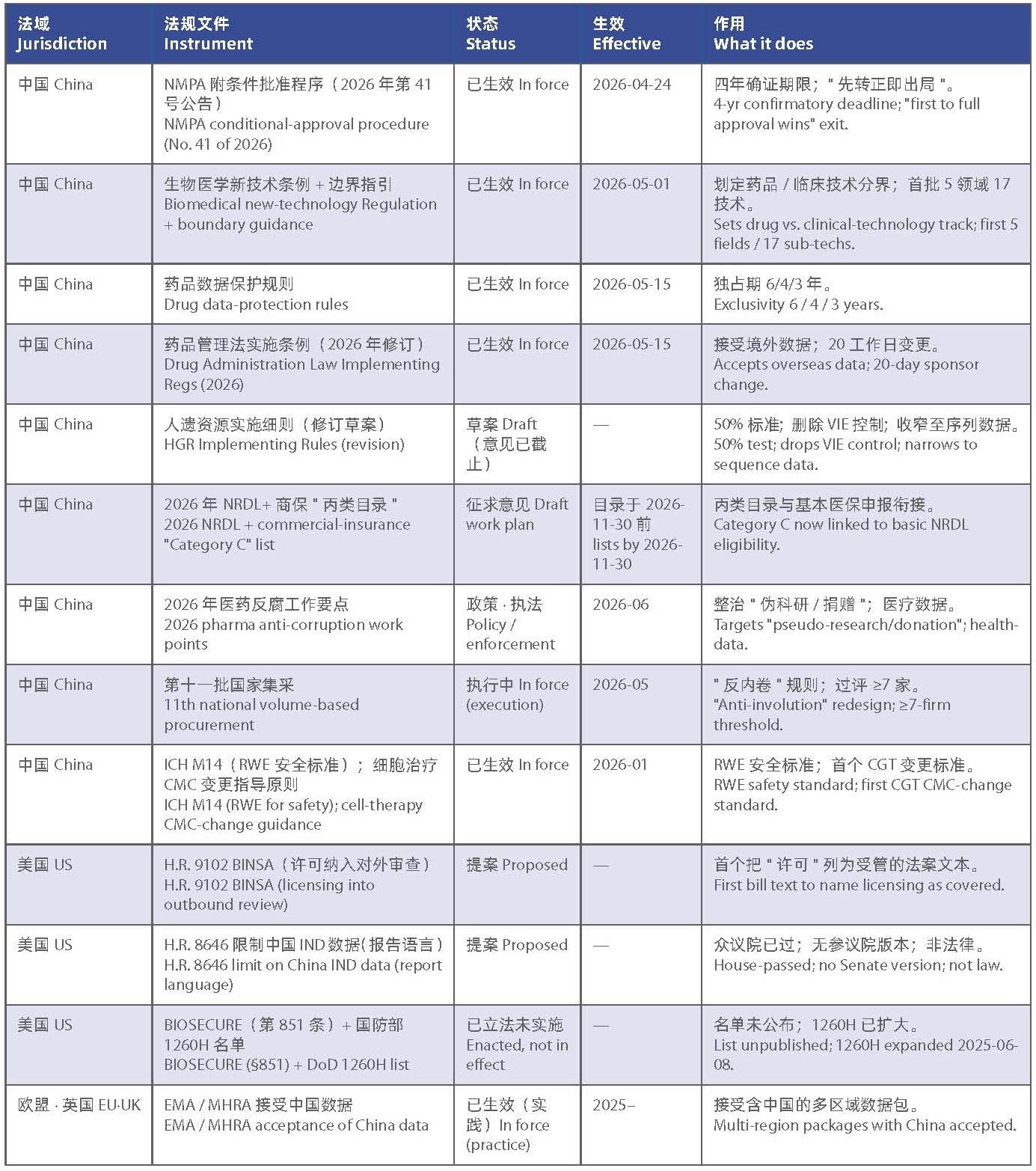
方达律师事务所 生命科学与医疗健康 — 监管与市场观察 第 01 期 | 2026 年 6 月(涵盖 2026 年 4 月中旬 – 6 月中旬)
定期梳理对生命科学与医疗企业、投资者及其顾问具有意义的跨境法律、监管与商业动态,覆盖中国内地、香港、美国与欧盟。
本期要目
- 中国重写附条件批准:四年内完成确证研究,率先获得常规批准者胜出。
- 中国首部国务院级“生物医学新技术”条例生效,由此决定产品按药品还是按临床技术监管。
- 《药品试验数据保护实施办法》于 2026 年 5 月 15 日生效,制定药品数据保护与境外数据领域新规则。
- 中国正在放宽人类遗传资源(HGR)规则,但目前仍为草案。
- 医保目录 2026 年调整启动,商保“丙类目录”正式与基本医保目录衔接。
- 十四部门确定 2026 年医药反腐重点,新增整治“伪科研”、“伪捐赠”形式的带金销售。
- 美国法案(H.R. 9102)首次拟将“许可”本身纳入对外投资审查,目前为提案阶段,尚未立法。
- 美国众议院推动限制中国临床数据,欧盟与英国则维持接受态度。
要闻
1. 中国重写附条件批准规则
- 状态: 已生效 | 中国 | 国家药监局2026年第41号公告,2026年4月24日起施行
- NMPA 全面修订了附条件批准程序。附条件批准的药品须在四年内完成确证性研究,注册证有效期亦与该期限挂钩。更值得关注的是,一旦同靶点、同适应症的同类药品获得常规批准,其余附条件品种即丧失资格。同时,附条件品种也不能作为仿制药的参比制剂。
- 关注要点: 附条件批准是重症药物在华上市的主要加速通道,新规影响范围覆盖所有计划在华注册的企业。四年期限将倒逼企业重新排布研发节奏,而“谁先转正谁留下”的退出机制意味着,抢先是生存所需,而不再是申报策略上的锦上添花——同靶点、同适应症的同类药品若先完成确证,持有人即可能丧失附条件资格。鉴于该情况直接涉及监管与上市规划、竞争定位与资产估值;在交易中,须将相关风险纳入尽职调查范围,并于开发承诺中予以充分反映。
2. 中国首部国务院级“生物医学新技术”条例生效
- 状态: 已生效 | 中国 | 国务院《生物医学新技术临床研究和临床转化应用管理条例》,2026年5月1日起施行;国家卫生健康委《生物医学新技术与药品、医疗器械界定指导原则(暂行)》,2026年5月1日起施行
- 中国首次在国务院层面为“生物医学新技术”立规,首批覆盖五大领域、十七项子技术,其中尤以细胞、基因与体细胞等技术领域最受业界关注。条例已于 2026 年 5 月 1 日施行;卫健委 4 月 30 日的配套文件划定了“生物医学新技术”(由卫健委按临床应用监管)与“药品/医疗器械”(由 NMPA 按注册监管)的监管边界。条例同时要求严格知情同意、禁止向受试者收费,并对健康损害责任与数据/隐私保护作出明确规定。
- 关注要点: 对于任何在中国开发或商业化细胞、基因及其他新型技术产品的企业、医院或研究机构而言,这一规定解决了一个根本问题:到底走药品路线(NMPA注册),还是走临床技术路线(卫健委审批)?这个选择将直接决定审批路径、谁有资格作为申办方,以及临床实施和数据合规方面的各项义务,包括知情同意、不得向受试者收费、责任承担和隐私保护等。本领域内的任何合作、供应或技术转让交易架构,也都必须以此为基础来设计。
3. 中国药品数据保护与境外数据规则已生效
- 状态: 已生效 | 中国 | 2026年5月15日起施行
- 2026 年 5 月 15 日,NMPA 的药品数据保护规则正式生效,建立了分层独占期制度。据现有报道,创新药(第1类)与境外首发原研药可享有6年独占期,改良型药为4年,首仿申请人为3年;在此期间,未经持有人同意,NMPA不会批准依赖受保护数据的改良型、仿制药或生物类似药。同日施行的《药品管理法实施条例》2026 年修订,则首次在法规层面明确规定:境外临床数据“符合国务院药品监督管理部门有关规定的”,可用于中国药品注册申请;具体技术标准仍沿用 2018 年发布的《接受药品境外临床试验数据的技术指导原则》,总体要求数据真实、完整、准确、可溯源,能够支撑有效性与安全性评价,符合 ICH GCP 与药品注册检查要求,且不存在影响有效性、安全性的种族敏感性因素。修订还将申办方变更审查时限统一规定为 20 个工作日。
- 关注要点: 对市场而言,影响是双向的。对原研方,中国注册资料现具有明确的独占期,构成产品生命周期与市场保护策略以及资产估值的实质考量。对仿制药或生物类似药开发者,则是依赖受保护数据时的时间与同意约束。对引入境外产品者,接受境外临床数据可加快其在华注册。任何中国法分析前,请先核实相关规定的现行文本。
4. 中国正在放宽 HGR 规则——但目前仍是草案阶段
- 状态: 征求意见稿 | 中国 | 2026年5月8日发布,2026年6月7日意见反馈截止
- 2026年人类遗传资源实施细则草案拟在多处放宽现行规定:“外方”认定将以 50% 股权或表决权为界,并取消此前可能套住 VIE 结构的“实际控制”概念;“人遗信息”将向基因序列数据收窄,明确不含单纯的临床、影像、蛋白与代谢数据;对不涉及人遗信息出境的合作,还将增设快速备案。但上述修订均尚未成为正式法律。在此之前,仍以 2024 年修订的人遗条例与 2023 年 7 月 1 日施行的实施细则为准;而中国法规草案在征求意见至定稿之间往往会有所收紧,建议持续关注后续进展。
- 关注要点: 人遗规则触及任何涉及中国人类遗传样本或数据的活动,譬如临床试验、产学研合作、CRO 安排与跨境数据流动。一旦定稿放宽,将广泛降低国际研发的现实困难。在此之前,应把人遗审批/备案作为这些活动的现实合规关口。按现行规则规划;合同涉及数据流转时,宜约定以最终文本而非草案为触发条件。
5. 2026 医保目录调整启动,商保“丙类目录”与基本医保目录衔接
- 状态: 征求意见稿 | 中国 | 《2026年国家基本医疗保险、生育保险和工伤保险药品目录及商业健康保险创新药品目录调整工作方案(征求意见稿)》等文件,2026年5月9日发布,2026年5月15日意见反馈截止,最终目录于2026年11月30日前公布
- 国家医保局开启 2026 年国家基本医保药品目录(NRDL)调整工作,同步开展商业健康保险创新药品目录(“丙类目录”,已进入第二年)调整。本轮调整首次将“已纳入 2025 年丙类目录”列为申报基本医保目录的条件之一,正式衔接两个目录。本轮亦向 2023–2026 年由附条件转为常规批准的药品开放申报。目前仍属征求意见阶段的工作方案;最终目录预计于 2026 年 11 月 30 日前公布。
- 关注要点: 医保报销是创新药在华商业价值的核心驱动,因此2026年的调整结果关系到任何厂商的定价、上市与收入规划,以及投资者据此建立的资产模型。新的衔接使商保丙类目录成为通往基本医保目录的过渡通道。应将其纳入准入策略;并请留意,方案目前仍处征求意见阶段,最终目录于 11 月 30 日前公布。
6. 十四部门定下 2026 年医药反腐重点
- 状态: 政策/执法重点(非强制性) | 中国 | 国家卫健委等14部门《2026年纠正医药购销领域和医疗服务中不正之风工作要点》,2026年6月8日印发
- 以卫健委牵头的十四个部门联合确定了 2026 年纠正医药购销和医疗服务中不正之风的工作要点,继续深化集中整治。今年的新增重点领域包括:整治以“科研”、“捐赠”为名的变相带金销售(“伪科研、伪捐赠”),以及医疗数据管理。需要说明的是,上述要点属于指导执法的年度工作政策文件,并非具有约束力的法律法规。
- 关注要点: 对任何在华有商业运营或与医务人员互动的企业而言,这是现实的合规与执法风险。将“伪科研”、“伪捐赠”渠道纳入重点,直接关系到医学事务、研究者发起研究、资助、捐赠与演讲/咨询安排如何开展——故无论是否涉及交易,均关乎内控、监测与风险敞口。涉及交易时,则对合作方的推广行为提出更高要求,支持强化反贿赂陈述、审计权与终止触发条款。
7. 美国法案首次把“许可”列为对外审查触发点——H.R. 9102(BINSA)
- 状态: 拟议(已提出,尚未成为法律) | 美国 | 《生物技术投资国家安全法案》(Biotech Investment National Security Act of 2026, BINSA Act),H.R. 9102,2026年6月2日提出
- 将企业层面的对外许可(out-licensing)纳入美国对外投资审查范围的立法尝试,已从信函提议阶段推进至正式法案文本阶段。《2026年生物技术投资国家安全法》(H.R. 9102)拟将“生物技术”增列入对外投资审查规则(即COINS框架)的覆盖行业,并据称系联邦法案首次将“向受管辖外国主体许可一项受禁技术”列为受管辖交易。该法案要求财政部对涉及知识产权或药物发现平台许可的交易给予“特别审议”,相关规定将于法案颁布后一年生效。就现行制度而言,尚无美国法规直接管辖单纯的对外许可:CFIUS审查入境并购,对外投资规则针对股权投资。本次法案旨在弥合这一监管缺口——但目前该法案仅处于提出并提交委员会阶段,尚未经委员会审议,亦无参议院对应版本。
- 关注要点: 此事关系到任何投资中国生物技术或自其引进的美国或跨国企业,尤其是许可交易的跨境结构如何规划——不仅限于正在推进中的某笔交易。当前尚无需为并不存在的审批环节预作定价;但在搭建新的涉华安排时,在协议中预先设置明确的“法律变更”条款(包括由谁通知、谁承担申报义务、若日后对外审查延及许可则未付里程碑款项如何处理等),可视为一项成本相对低廉的风险对冲安排。相关风险主要暴露于美方或跨国企业一方,以及其设立的美国NewCo或SPV。
8. 美国推动限制中国临床数据,欧盟与英国仍接受
- 状态: 拟议(美国) | 已生效(欧盟、英国)
- 美国方面,众议院于2026年6月4日以213票对210票的微弱优势通过2027财年农业-FDA拨款法案(H.R. 8646)。其中要求FDA在IND阶段不得“接受、审查或考虑”来自中国(以及伊朗、朝鲜、俄罗斯)研究机构的临床数据;但该要求目前仅载于委员会报告而非法案正文,因此不具法律约束力,且参议院尚未提出对应版本,暂无可对接的立法进程。与此同时,欧盟与英国仍接受将中国数据作为多区域临床试验的一部分:serplulimab即凭借全球性ASTRUM-005试验在两地获批用于一线治疗广泛期小细胞肺癌;MHRA首席执行官亦公开表示该机构属于“国际互信生态”的一部分。尚待观察的是,欧盟与英国的接受态度是否会延伸至“仅来自中国”的单一区域数据,而不限于设有非中国组别的多区域试验。
- 关注要点: 此事影响任何开展或依赖中国临床数据的主体的全球开发与监管策略,包括试验设计、注册排序,以及价值在何处实现等,而非仅及某笔交易如何分配风险。应将美国相关措施视为一项具体、可预见、与 FDA 数据接受挂钩的法律变更,而非简单的开关;并设计试验与地域策略,使美国路径收窄时欧盟/英国(及其他 ICH)的价值仍存。
其他关注
- 中国: 关于第十一批国家集采,国家医保局已于2026年5月22日发布公告,宣布第十一批国家集采进入执行阶段。这是首次以“反内卷”为理念优化规则运行的集采项目。通过设锚点价、复活机制、按厂牌报量、过评企业不少于七家等设计,标志着从纯最低价竞争模式转向,有望温和重塑专利到期后的市场格局。 关于技术指导原则,NMPA发布了适用ICH M14的公告(2026年第16号公告),用于真实世界证据在药品安全性评估中的应用;CDE则发布了细胞治疗药品药学(CMC)变更技术指导原则(试行)(2026年第13号公告),为CAR-T、TIL、干细胞等细胞治疗产品的生产变更提供了首个全生命周期标准。上述两项指导原则均直接关系到企业的药物警戒义务与CMC/变更控制实务。
- 美国/全球: 关于BIOSECURE,第851条虽已正式立法,但尚未进入实施阶段。目前既无受关注实体名单,也无采购规则修订;既有合同另享五年过渡期(安全港)。值得留意的是,国防部1260H名单已于2025年6月8日完成扩容(新增Novogene与药明康德),而一旦BIOSECURE正式生效,被列入1260H名单的企业将自动被认定为“受关注公司”。需特别注意的是,该法本质上属于采购法范畴,并非交易禁令,其约束力仅通过美国政府合同的履行环节传导至私主体层面,企业间自主交易本身并不受直接限制。 关于出口管制,目前暂无新动向。美国商务部工业与安全局(BIS)针对部分生物技术设备(ECCN 3A069/3E069,自2025年1月起生效)对华适用拒绝推定原则;中国方面,2026年《两用物项出口管制目录》及2023年版目录均涵盖人类生殖细胞编辑与克隆技术相关物项。上述管制措施均直接关系到相关设备或技术方法能否实现跨境流转,是交易可行性评估中不可忽略的考量要素。
法规动态追踪
本期专题 一图看懂涉及对外许可的四种美国国家安全工具
与中国生物技术有关联的企业,如今面对四种各自独立的美国国家安全工具,每一种触及画面的不同部分。CFIUS 审查“进来的钱”——对美国企业的投资与收购,而不是“出去的许可”。对外投资规则(COINS/OISP)触及“出去的股权”——流向中国特定科技行业的股权,但目前不含生物技术。BIOSECURE 触及联邦采购,只经由美国政府合同的履行才触及私主体,且尚未生效。而 H.R. 9102(BINSA)是拟议中的“第四块”,也是唯一直接点名“许可”的:其条文要把“向受管外国主体许可一项受禁技术”纳入对外审查。
务实的结论是:就目前而言,似乎还没有哪个美国法规能直接管到一笔单纯的对外许可,而最有可能改变这一局面的,正是眼下已经落到条文层面的这项法案。因此,对于任何正在搭建或持续维系涉华生物技术合作关系的企业来说,稳妥的做法并非为一个尚不存在的审批关口预先设置条件,而是在当前的协议中就嵌入一套清晰的“法律变更”机制:界定清楚触发情形;明确一旦日后对外审查扩大至许可交易,由谁承担申报义务和配合责任;以及约定如果未来申报被否决或附加了条件,尚未支付的里程碑款项该如何处理。还有一个结构层面的问题值得提出来:界定所谓“中国境外NewCo”的那层股权安排,恰恰是未来对外审查最有可能抓住的切入点。因此,选择什么样的交易架构,本身就是在选择由谁来承担监管风险。
对您的影响
- 重新审视加速通道资产的中国监管与上市策略——四年确证期限与竞品退出规则,使“第一个拿到批件”已经从申报节奏问题变成了竞争格局问题,不止是递交文件时该注意的细节。
- 对细胞、基因或其他新型技术,先对产品分类,即属于药品线还是生物医学新技术线,再确定开发、临床或合作计划。
- 把已生效的数据保护独占期与 2026年医保目录调整(含商保丙类目录)纳入定价、市场准入与生命周期规划;投资者应在资产模型中反映这两项因素的影响。
- 对照当前“伪科研/伪捐赠”的执法重点,尽早对在华合规体系做压力测试,覆盖医学事务、资助、捐赠及医务人员互动等环节。不管眼下有没有交易在进行,这项工作都宜先行。
- 凡是涉及中国人类遗传资源样本或基因数据的项目,都应将人类遗传资源审批/备案视为一道现实的准入门槛,按现行规则制定计划,同时密切关注相关条例修订草案的动态。
- 对美方面,持续跟踪涉华数据相关的监管变化,同时保留美国以外(欧盟/英国/ICH)的替代选项;在搭建跨境生物技术交易架构时,可先行设计好针对对外投资审查变化的应对条款(尤其留意H.R. 9102的走向),并对交易对手方做BIOSECURE / 1260H名单相关的风险筛查。
本简报仅供一般参考之用,不构成法律意见,亦不建立律师与客户之间的委托关系。 其内容基于截至2026年6月的公开信息,可能因后续发展而发生变化。 如您就特定事项需要法律意见,请联系您在方达律师事务所的联系人。 由方达律师事务所生命科学与医疗健康团队编制。



